Formulae
of molecules and atoms (radio spectra)
Meaning of quantum numbers
and related symbols
(Most contents from NIST diatomic spectral database documents)
I or Ii ¨C Angular momentum quantum number of nuclear spin for one (or ith) nucleus
S ¨C Resultant angular momentum quantum number of electron spins
¦˛ ¨C Projection of S on the molecular axis (for HundˇŻs case a only)
¦« ¨C Absolute value of the projection of the resultant orbital electronic angular momentum on the molecular axis (for HundˇŻs case a and b only)
¦¸ ¨C Absolute value of the projection of the total electronic angular momentum on the molecular axis (= ¦˛+¦«)
N ¨C Rotational angular momentum quantum number, excluding electronic and nuclear spins
J ¨C Resultant total angular momentum quantum number, excluding nuclear spins
mJ ¨C Projection of J along any given axis
Fi ¨C Resultant angular momentum quantum number including nuclear spin for i nuclei
Itot ¨C Resultant angular momentum quantum number of two or more nuclear spins that couple together before being coupled into total rotational angular momentum F.
F ¨C Resultant total angular momentum quantum number
mF ¨C Projection of F along any given axis
X ¨C Quantum number employed when FiˇŻs are not good quantum numbers. This value simply numbers the levels from lowest to highest energy for the same F quantum number
v ¨C Vibrational quantum number
¦Íi ¨C vibrational quantum numbers for the ith vibrational mode for molecules with more than 2 nucei. Vibrational modes are usually sorted with increasing energy (with decreasing degree of symmetry) and designated as ¦Í1, ¦Í2, ¦Í3,ˇ sequentially.
l ¨C Quantum number for the projection of degenerate 2-dimensional bending vibration angular momentum on molecular axis (for linear molecules only)
¦˛, ¦°, ¦¤ -- Electronic state designation for which ¦« = 0, 1, 2, respectively. The symbols X, A, B, E, F, ˇ precede the state designation where X refers to the ground state, A refers to the first excited electronic level, ...
General Formulas
Radiative transfer
The general radiative transfer
equation for a ray is
![]() ,
,
where I¦Í is the
radiation intensity in ergs-1cm-2Hz-1sr-1,
s is the radiation transfer route length, ¦Ę¦Í is the absorption
coefficient (opacity) in cm-1, ¦Ĺ¦Í is the emission
coefficient (emissivity) in ergs-1cm-3Hz-1sr-1.
It is convenient to define an optical depth ¦Ó¦Í as
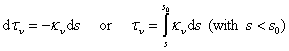 .
.
Note that both ¦Ó¦Í and s are positive quantities, while the minus sign in the first equation
means the two quantities increase in opposite directions, with ¦Ó¦Í =0 at s0. The radiation is going along increasing direction of s. Then,
the radiation
transfer equation becomes
![]() ,
,
with the source function
![]() .
.
In thermodynamic equilibrium
(TE), I¦Í= S¦Í= B¦Í(Tk),
with Tk being the kinematical temperature of gas (e.g., in
the interior of a star). Here, the Plank function is
![]()
For molecular gas not in TE but in local thermodynamic equilibrium (LTE), although the radiation is not in
equilibrium with the gas, we usually still can define an single excitation
temperature Tex for all energy levels of a molecule (according to Boltzmann distribution, see below), so that S¦Í=
B¦Í(Tex) can be
fulfilled for all transitions. Even when the molecular gas is not in LTE, this definition of Tex
still can be applied, but Tex may be different for different
transitions of the same molecule.
The formal solution of the
radiative transfer equation is
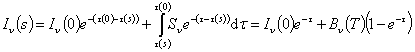
In the second equation, we have replaced the optical depth
difference ¦Ó(0)- ¦Ó(s) by a new optical depth ¦Ó that is defined to be zero at s=0 and
increases in the same direction as s. If we subtract the back ground intensity I¦Í(0)= B¦Í(Tbg) from the measured
intensity I¦Í(s), we get
![]() ,
,
or in terms of brightness
temperature
![]() .
.
Here, TR is the
brightness temperature directly measured by radio telescope using the beam
switch method (so that the back ground brightness has been subtracted). J(T)
is Relaigh-Jeans temperature:
![]()
Einstain coefficients Aul, Bul
and Blu
The three Einstein coefficients that describe the interaction between molecule and radiation are the spontaneous transition probability Aul in s-1, the induced emission or absorption probability Bul and Bul in s-1erg-1cm2sr. For gaseous molecule with radiative transitions between an upper level ˇ°uˇ± and a lower level ˇ°lˇ±, the three coefficients are defined by
![]()
Here, flu is oscillator
strength, e is electron charge, me is electron mass, c
is the speed of light, ¦Í is the transition frequency from upper, gu,
gl are statistic weights. Therefore, for gas number density nu
and nl in the two levels, the spontaneous photon emission
rate is nuAul (s-1) while the
induced photon emission or absorption rates in a radiation field with mean
radiation intensity of J(¦Íul) (erg/s/Hz/cm2/sr) are nuBulJ(¦Íul)
and nlBluJ(¦Íul) (s-1),
respectively. (NOTE1: The definition of Bul
and Blu may be slightly different by using radiation energy
density U(¦Íul) instead of the mean intensity J(¦Íul)
according to the relationship ![]() .) (NOTE2: If the levels are degenerate,
the above emission rates are only for transition from one initial sublevel to
all final sublevels. Therefore, to calculate the total emission rates, one must
multiply them by the statistic weight of the initial level.)
.) (NOTE2: If the levels are degenerate,
the above emission rates are only for transition from one initial sublevel to
all final sublevels. Therefore, to calculate the total emission rates, one must
multiply them by the statistic weight of the initial level.)
The Einstein coefficient Aul
is related to the electric or magnetic dipole moment ¦Ěul of the transition as
![]() .
.
For rotational transitions, the
dipole moment is usually expressed as
![]() ,
,
in which ¦Ěx is the permanent dipole moment along axis x (the symmetry
axis of a
molecule along which the permanent dipole moment is non-zero), xSul
is the so called line strength or transition strength for the
transition from level ˇ°uˇ± to ˇ°lˇ±. The line strength is only determined by the direction part of the molecular wave
function and so is only related to the symmetry property
of the molecule.
Especially, the line strength has no relation to molecular weight, size,
composition etc. The formulas of the line strength for molecules with different
symmetry types are given in detail in latter sections.
Absorption coefficients
in terms of Einstein coefficients
Absorption coefficient per unit volumn of
molecular gas is
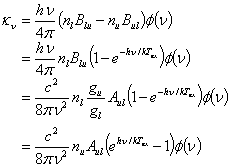 .
.
From here on, the subscript ˇ°ulˇ± of frequency has been omitted for clarity. Substituting Aul into this formula, one gets
![]()
Boltzmann distribution
In local thermal equilibrium
(LTE), the molecular energy level population distribution nj
obeys the Boltzmann distribution law
![]() ,
,
in which N is the total number
density, Tk
is the kinematical temperature, Ej is the energy of the level j, and Q is the
partition function which is defined as
![]() .
.
One must be careful about the calculation of Q when fine structure or hyperfine structure lines are considered. For those molecules with non-zero electron and/or nuclear spins, the (hyper-) fine-structure splitting increases the total number of levels and makes the Q value much larger. Therefore, with the same number density N, the population nj of each (hyper-) fine level becomes smaller, and the individual (hyper-) fine line intensity also becomes weaker. This consideration is also valid for those asymmetric top molecules that have many energy levels due to the lack of symmetry.
Relative intensity of optically
thin rotational lines
In the case of optically thin case, we can omit the opacity effect and use the spontaneous photon emission rate to roughly estimate the relative intensity of different rotational lines. For a homogeneous and optically thin molecular cloud, we can omit the opacity effect so that the integrated emission line intensity just equals to
![]()
Here, the formulas of the Einstein A coefficient and the boltzmann
distribution law have been applied. With the formulas for pure rotational
transitions given in latter sections: ![]() and
and![]() , one has
, one has
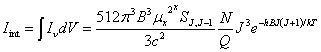
The strongest line occurs at![]()
![]() . Take the example of CO in the ground vibration state,
B=57635.968 MHz from CDMS database. Therefore, the strongest optically thin is
at
. Take the example of CO in the ground vibration state,
B=57635.968 MHz from CDMS database. Therefore, the strongest optically thin is
at![]() . For a temperature of T = 9.375 K, the strongest line is at
around J=2. For T = 300 K, the strongest line is around J=12.5. These results qualitatively
agree with the line intensity plots from CDMS database (see in the figures
below).
. For a temperature of T = 9.375 K, the strongest line is at
around J=2. For T = 300 K, the strongest line is around J=12.5. These results qualitatively
agree with the line intensity plots from CDMS database (see in the figures
below).

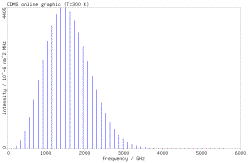
Rotation diagram
analysis
Although further extension of above discussions of relative intensity of optically thin lines just results in the formula for rotation diagram analysis (See for example the derivation by Cummins et al., 1986, ApJS, 60, 819), here I would like to restart from the formal solution of radiative transfer equation to derive it. From the formal solution of RT equation, the observed radio line intensity after the removal of back ground emission is
![]() .
.
Here, Rayleigh-Jeans approximation has been applied to Tex and Tbg. In the optically thin case,
![]()
From above discussions of absorption coefficient, one has the optical thickness of a homogeneous cloud as
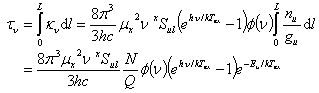 .
.
Here, L is the size of the cloud, N is the total column number density of the molecules and Q is the partition function. Substituting it into previous formula and integrate over frequency, one gets the integrated line temperature
![]()
By using the relation ![]() and applying
Rayleigh-Jeans approximation, we have the integrated line intensity
and applying
Rayleigh-Jeans approximation, we have the integrated line intensity
![]() .
.
Then we obtain the standard formula for rotation analysis:
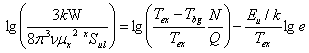
(See the similar derivation by Kawaguchi et al., 1995, PASJ, 47, 853) Note that this formula is applicable only for energy levels (in unit of K) far lower than kinematical temperature of the gas. Otherwise, non-LTE effects may emerge.
Energy level balance
equations (Rate equations)
In above discussions, TE or LTE has been assumed. However, the actual molecular gas may not fulfill that condition. For example, when external radiation field plays an important role in the excitation of energy levels, LTE is usually not met. In this case, one needs to compute the level populations by the level balance equations:
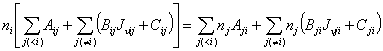
Here, Aij and Bij are Einstein constants, Cij is collision rate in unit of s-1, J¦Íij is the mean radiation intensity in unit of Wm-2Hz-1Sr-1 that must be determined by solving radiative transfer equation. This set of equation must be closed by the equation of particle conservation
![]()
Here N is total number density of the species. Once the mean radiation intensity is known, these linear equations of level population can be easily solved by standard method.
Format of molecular
energy level specification
Similar as atomic energy level formulas, molecular formula also take the form
![]()
Here the terms before the parenthesis is electronic state specification. X means it is in the ground electronic state (n=0). X may be replaced by A, B, C, ˇ for excited electronic states with n=1, 2, 3, ˇ However, molecules in electronically excited states are rare, because the huge excitation energy usually will get the molecule dissociated. In the second electronic term, ¦« represents projection of resultant electronic orbital angular momentum along the major symmetry axis of the molecule. Upper case Greek letters ¦˛, ¦°, ¦¤, ˇ are used for |¦«| = 0, 1, 2, ˇ respectively. However, for strongly asymmetric molecules for which there is no obvious symmetry axis, the Greek letters may be replace by upper case alphabets A, B, E, F, ˇ to indicate the different symmetry properties. S is the total electron spin quantum, hence 2S+1 is also called multiplicity of each electronic level. ¦¸ represents the resultant projection of total electronic angular momentum along the major symmetry axis of the molecule (mainly composed of projection of electronic spin ¦˛ and that orbital angular momentum ¦«, but in the case of bending linear molecules, it also include the projection of the 3D bending angular momentum l).
The quantum numbers (Qn) in the parenthesis are related to vibration
and rotation of the molecule. Those Qns not related to spin are usually called
primary Qns, while those involving spins are called secondary Qns. The Qn v is for
vibrational states of diatomic molecules, while ¦Íil is used for linear chains
where i = 1, 2, 3, ˇ for different vibrational frequency modes ¦Í1, ¦Í2,
¦Í3, ˇ. The superscript l
is for the projection of the 3D vibrational angular momentum along the symmetry
axis. N is the Qn for rotational angular momentum, J is the Qn for resultant
total angular momentum (including electronic components), ¦¸ is the Qn for
resultant total angular momentum in the HundˇŻs case (a) coupling, ¦« is the Qn
for projection of resultant total angular momentum along symmetry axis. The rest
are secondary Qns: F1, F2,
ˇFi, ˇ are for resultant angular momentum when the ith non-zero nuclear spin is taken into
accound; Itot is the
resultant of nuclear spins of two identical nuclei that usually couple first
before being coupled to the total angular momentum; F is for the ultimate total angular momentum (the only quantity
that strictly conserves).
Atoms
(Most contents from chapter 5 of Townes C.H. and Schawlow A.L., Microwave Spectroscopy, New York: McGraw-Hill, 1955)
Quantum numbers
Usually, only hyperfine structure transitions of atoms appear in millimeter wavelength. Quantum numbers for an atomic level with nuclear spin are L, J, mJ, I, F, mF.
Energy level
There
are mainly two kinds of hyperfine structure splitting due to electric
quadrupole momentum and magnetic
dipole momentum respectively.
The formula of quadrupole hyperfine
structure level is
![]()
With
![]()
Here, eqJQ is the quadrupole
coupling constant.
The
formula of magnetic dipole hyperfine structure level is
![]()
Here, a is the so-called interval factor, a constant for a given electronic state and nucleus.
With external magnetic field, ˇˇ(Zeeman effects)
Statistic weight
For the pure hyperfine structure level: SF = 2F+1
Selection rules
Only pure hyperfine transitions fall in microwave. The selection rule for these pure hyperfine transitions is
![]()
Magnetic dipole moment
for the transitions
The pure hyperfine transitions in microwave are usually only strong
in the ground electronic state 2S1/2 of atoms, because
most atoms in space are populated in this ground level and the hyperfine
structure splitting is the largest in this state. For pure hyperfine
transitions in the 2S1/2
state with external magnetic filed, the magnetic dipole moment for
transition ![]() is:
is:
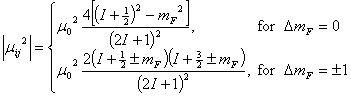
Here, mF is the projection of total angular momentum F
along the direction of external magnetic filed B, ¦Ě0 = he/4¦Đmc is the Bohr magneton (the
statistic weight for each non-degenerate magnetic level is 1). Note that the transitions
with ![]() are always
polarized with their magnetic vector parallel to magnetic filed B, while
transitions with
are always
polarized with their magnetic vector parallel to magnetic filed B, while
transitions with ![]() are always
polarized with their magnetic vector perpendicular to magnetic filed B.) The
line strength in this case is
are always
polarized with their magnetic vector perpendicular to magnetic filed B.) The
line strength in this case is
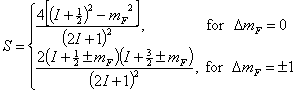
For pure hyperfine transitions without
external magnetic field, the magnetic dipole moment averaged over all
transitions with the same ¦¤mF
and summed over 3 directions for absorption transition ![]() is
is
![]()
The line strength in this case is
![]()
Inversely, the magnetic dipole moment for emission transition ![]() should be
should be
![]() ,
,
Here, gF = 2F+1 = 2(I+1) is
the statistic weight of each level.
Miscellaneous
l
Angular momentum of an electron: ![]()
l
Magnetic moment of an electron: ![]() (¦Ě0
is the Bohr magneton,
(¦Ě0
is the Bohr magneton, ![]() is the fine
structure constant.)
is the fine
structure constant.)
l
Angular momentum of a nucleus: ![]()
l
The most famous example of pure hyperfine structure
transition is the 21cm H emission line (1.420405 GHz) from
the interstellar space of the Galaxy.
Diatomic
molecules
(Most contents from chapter 1 of Townes C.H. and Schawlow A.L., Microwave Spectroscopy, New York: McGraw-Hill, 1955)
Quantum numbers (my
opinion)
For simple diatomic molecules: J(=N), v (e.g., X1¦˛ state molecules)
with only fine structure splitting: N, v, J (e.g., X3¦˛ state molecules)
with only hyperfine structure splitting: J(=N), v, F1, F
with both fine and hyperfine structure splitting: N, v, J, F1, F
for HundˇŻs case a coupling (L>0,S>0): ¦¸, ¦«, J (e.g., X2¦° state molecules)
for HundˇŻs case a with hyperfine structure: ¦¸, ¦«, J, F1, F (e.g., OH)
Here, N, v, ¦¸, ¦« are called principal quantum numbers, J, F1, F are called spin involved quantum numbers. For molecules with identical nuclei, the sign of ¦« should reflect parity. F1 is needed only when both nuclei have non-zero spin. In the case where J=N, the convention is to use J and omit N.
Energy level
For a rigid rotor with neither electronic nor nuclear spin, level energy
![]() ,
,
where h is PlankˇŻs constant, I is the momentum of inertia, B is rotation constant, J is the rotational quantum number.
For a non-rigid rotor, if
we adopt Morse potential ![]() (where D is the dissociation energy of the molecule, re is equilibrium distance
between nuclei, a is a constant) for low vibration states with r ~ re,
the level energy containing centrifugal stretching is
(where D is the dissociation energy of the molecule, re is equilibrium distance
between nuclei, a is a constant) for low vibration states with r ~ re,
the level energy containing centrifugal stretching is
![]()
Here, J and v are rotation and vibration quantum numbers, subscript e means quantities when atoms are in equilibrium position, and
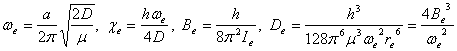
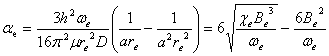
Here, ¦Ě=m1m2/(m1+m2) is the reduced mass the two nuclei m1 and m2.
A more appropriate solution for any potential function that can be expanded as a series of power of (r-re) in the neighborhood of the potential minimum is the DunhamˇŻs solution:
![]()
Here, l and j are summation indices, Ylj are coefficients depending on molecular constants. In the case of Be/¦Ře<<1 (say, for molecules not very light, e.g., the molecule H2 is a bad case), one has the approximate relations:
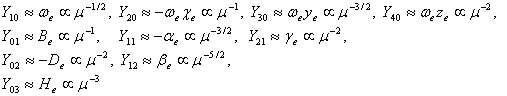
(Note: Part of the scaling relation is from the documents of NIST diatomic spectral database.) The above scaling relations with the reduced mass ¦Ě of the molecule are very useful to approximately calculate the energy levels of rare isotopic diatomic molecules from their main isotope, because for many molecules, the only significant difference among isotopomers is nothing but the reduced mass.
When electronic orbital angular momentum and/or spin exist, the energy level structure is determined by the angular momentum coupling schemes. There are four typical coupling schemes: the HundˇŻs cases (a,b,c,d) (see Appendix B for details). The most useful one is the HundˇŻs case (b) coupling, the case (a) occurs sometime, while the cases (c) and (d) are very rare.
For HundˇŻs case (a) coupling, the energy level is
![]()
Here Bv is the rotation constant corresponding to vibrational state v, A is also a rotation constant that is usually much larger than Bv. ¦¸ is the quantum number for the projection of J along the molecular axis.
For HundˇŻs case (b) coupling, the energy level is
ˇ.
For HundˇŻs case (c) coupling is identical to that of case (a). For HundˇŻs case (d), the energy level is
![]()
For fine
and hyper fine splitting, the energy levels areˇ (????)
What is the role of parity in the energy level
formula? (For example, the energy levels of SiC are different for different
parities.)
With external magnetic field, ˇˇ(Zeeman effects)
Statistic weight
For pure rotational levels: gJ = 2J+1
The only angular momentum not included in the quantum number J is nuclear spin. For rotational levels with non-zero nuclear spins (hyperfine structure splitting), the statistic weight for a pure hyperfine splitting level is
![]() ,
,
while that for a pure rotational level (not differentiating hyperfine components) is:
![]() .
.
ItˇŻs not difficult to verify the second equality using the quantum
angular momentum coupling rule:![]() .
.
Selection rules
Selection rules for electric dipole transitions in the absence of external field: (from NIST diatomic spectra database documents)
For X1¦˛ molecules: ![]()
For X3¦˛ molecules: ![]() (e.g., O2,
SO)
(e.g., O2,
SO)
Actually ![]() is also allowed
for intermediate coupling case,
is also allowed
for intermediate coupling case,
for magnetic dipole transitions: ![]()
For X2¦° molecules:![]() (HundˇŻs case a, e.g., NO)
(HundˇŻs case a, e.g., NO)
Electric dipole moment
for transitions
For the case of L=0, S=0 (say, 1¦˛ state),
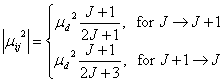
Here, ¦Ěd is the permanent electric dipole moment of the molecule. The line strength thus is
![]()
For the case of L>0 (nonzero electronic orbit momentum), the molecule can be treated in analogue of symmetric top, so that the rotational line strength for HundˇŻs case (a) coupling is
![]() ,
,
in which ¦« is the projection of L along the molecular axis. For the HundˇŻs case (b), the line strength is
ˇ.???????
![]() ??????
??????
In the case of fine structure splitting (S>0), the total of line strength of all fine structure lines
just equals to the rotational line strength given by above formulas with the
quantum number J replaced by N, while the relative intensity (RI) of
fine structure lines should be determined by the quantum angular momentum
coupling formula (see details on this in Appendix A). Eventually, the line strength
for a single fine structure transition is simply S(N,J) = S(N) * RI.
Linear molecules
(Most contents from chapter 2 of Townes C.H. and Schawlow A.L., Microwave Spectroscopy, New York: McGraw-Hill, 1955)
Quantum numbers
For the case with L=0,S=0,Ii=0: J, ¦Í1,ˇ, l (J=N)
with L=0,S>0,Ii=0: N, ¦Í1,ˇ, l, J
with L=0,S=0,Ii>0: J, ¦Í1,ˇ, l, F1,ˇ, Itot, F (J=N)
with L>0,S>0,Ii>0: N, ¦Í1,ˇ, l, J, F1,ˇ, Itot, F (HundˇŻs case b)
with L>0,S>0,Ii=0: ¦¸, ¦«, J (HundˇŻs case a, e.g., X2¦° state molecules)
with L>0,S>0,Ii=0: ¦¸, ¦«, J, F1,ˇ, Itot, F (HundˇŻs case a)
l Here, N, ¦Í1,ˇ, l, ¦¸, ¦« are called principal quantum numbers, J, F1,ˇ, Itot, F are called spin involved quantum numbers. For molecules with identical nuclei, the sign of ¦« may reflect parity. F1,ˇ are needed only when any nuclei have non-zero spin. Itot is needed when two spins couple with each other before being coupled to the total angular momentum.
l There are two ways to specify the vibration quantum numbers ¦Í1,ˇ values: 1) list all ¦Í1l values, with non-zero l as superscript: ¦Í1, ¦Í2l, ¦Í3,ˇ (suitable for small molecules, e.g., 0110 state of CO2); 2) only specify the non-zero ¦Í1l values (suitable for large molecules, e.g., ¦Í7=11,N=23,J=47/2 of C4H).
Energy level
With centrifugal stretching effects, energy level of a linear molecule is:
![]() with
with ![]() ,
,
where ¦Ái is rotational constants that reflect the change of B from its equilibrium value Be due to ith vibration mode. di is the degeneracy of the ith vibration mode, or the number of degenerate modes with the same ¦Ái, D is the centrifugal stretching coefficient.
For a degenerate excited bending mode, with the l-doubling considered, the energy level for the case of |l|=1 is
![]()
with
![]()
Here, ¦Ík is the zero energy of the ground level of the degenerate vibration mode, l is the bending vibration angular momentum quantum number, ql is the l-doubling coefficient. For higher excited bending modes with |l|>1, the l-doubling is usually very small (not important for mm wavelength).
With external magnetic field, ˇˇ(Zeeman effects)
Statistic weight
For pure rotational levels: gJ = 2J+1
As long as the molecule is not in the degenerate excited bending
mode, the only angular momentum not included in the quantum number J is nuclear
spin. For rotational levels with non-zero nuclear spins (hyperfine structure
splitting), the spin statistics is (2I+1). Therefore, the statistic weight of a
hyperfine is: gF = (2I+1)(2J+1) =2F+1.
Selection rules
Selection rules for electric dipole transitions in the absence of external field:
![]()
Electric dipole moment
for transitions
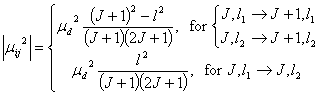
Here, ¦Ěd is the permanent dipole moment of the molecule and l1 and l2 represent the two l-doubled sublevels with lower and higher energy, respectively. One can easily see that for ground vibrational level with l=0, the formula is identical to that of the diatomic molecule. The line strength thus is
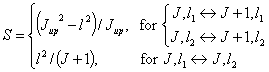
One can see the similarity of above formula with the dipole moment of symmetric top. Actually the excited bending mode of linear molecule can be considered as an asymmetric top with slight asymmetry. The quantum numbers of the excited bending mode can be analogously written in the form of asymmetrical top as:
For the higher l-doubling level: J,l => J|l|,J+1-|l|, and
For the lower l-doubling level: J,l => J|l|,J-|l|
Then, in the
slight asymmetry limit, the dipole formula is the same as that of a symmetric
top.
Symmetric top
(Most contents from chapter 3 of Townes C.H. and Schawlow A.L., Microwave Spectroscopy, New York: McGraw-Hill, 1955)
Energy level
For a rigid symmetric top, the energy level is
![]()
Here C is the rotation constant along the symmetry axis, B perpendicular to the axis. With centrifugal stretching considered, the energy level is
![]()
For a rigid symmetric top with l-type doubling, the energy level is
![]()
Here, |ς| ˇÜ 1 is a coefficient describing how well the bending motion is perpendicular to the molecular axis C, with a value of 1 for the entirely perpendicular case.
With external magnetic field, ˇˇ(Zeeman effects)
Statistic weight
Usually, a symmetric top has at least 3-fold of symmetry (except the rear case of accidental symmetric top made from linear molecules). The statistic weight of a pure rotational energy level is gJ = 2J+1. With hyperfine structure splitting, the statistic weight then becomes gF = 2F+1. For a molecular energy level with hyperfine structure splitting but with the hyperfine structure transitions unresolved, the statistic weight of such an unresolved rotational level is composed of two independent parts: pure rotational levels weight gJ and the spin statistics weight S(J,K). Then, the total statistic weight of the unresolved rotational level is the multiplication of the two parts:
![]()
The spin statistic weight for any given rotational level J of a 3-fold symmetric top is
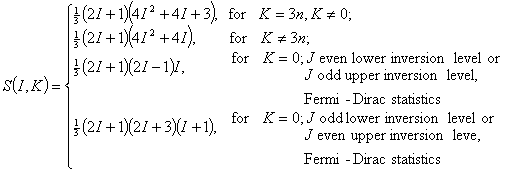
in which I is total nuclear spin quantum number, n is a non-negative integer. (A nucleus with odd mass number usually has half integer spin and so obeys Fermi-Dirac statistics, say, exchange of any pair of such identical fermion nuclei results in the reverse of the sign of the molecular wave function. In this case, one says the total wave function of the molecule is anti-symmetric w.r.t. the exchange of the identical nuclei. Oppositely, a nucleus with even mass number usually has integer spin and so obeys Bose-Einstein statistics, say, exchange of any pair of such identical boson nuclei will result in completely the same molecular wave function. One says that the total wave function of the molecule is symmetric w.r.t. the exchange of identical nuclei.)
Similarly, for a 4-fold symmetric top molecule, the spin statistic weight is
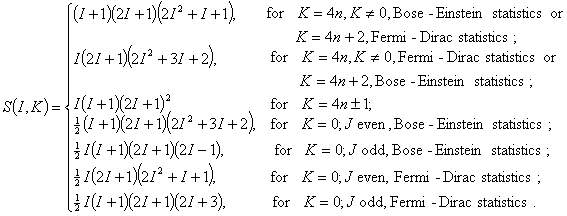
Selection rules
Selection rules for electric dipole transitions in the absence of external field:
![]()
Electric dipole moment
for transitions

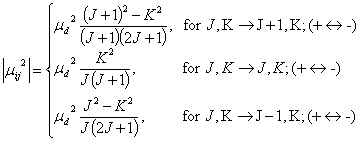
Here, ¦Ěd is the permanent dipole moment of the molecule. The J,K ¨C J,K transition is the inversion transition of molecules like NH3. One can easily see that for the limit case of K=0, the formula is identical to that of the diatomic molecule. The line strength thus is
![]()
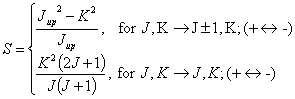 ?????
?????
One can easily see that the line strength is identical for both
emission and absorption. When (hyper-)fine structure splitting exists, the sum
of line strength of all (hyper-)fine lines should be just equal to the rotational
line strength calculated by above formulas. The relative intensity (RI) of each
(hyper-)fine lines should be computed according to quantum angular momentum
coupling formulation (see more details in Appendix A).
Asymmetric
top
(Most contents from chapter 4 of Townes C.H. and Schawlow A.L., Microwave Spectroscopy, New York: McGraw-Hill, 1955)
Energy level
For a rigid asymmetric top that is merely slightly asymmetric, the level energy is
For
prolate top: ![]()
For
oblate top: ![]()
in which A, B and C are rotation constants along the three molecular axes and the convention of A>B>C is assumed, and
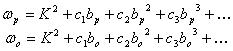
Here,
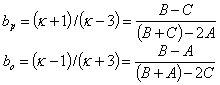
are asymmetry parameters for prolate and oblate tops, respectively, with
![]()
being KingˇŻs asymmetry parameter.
For a highly asymmetric rigid top, the energy level is
![]()
with ![]() only a function
of ¦Ę. A list of
only a function
of ¦Ę. A list of ![]() can be found in
G. Erlandsson, (1954, Arkiv For Fysik, 8, 189). There is a useful relationship:
can be found in
G. Erlandsson, (1954, Arkiv For Fysik, 8, 189). There is a useful relationship:
![]() .
.
For a non-rigid asymmetric top, the energy level including centrifugal stretching effects is
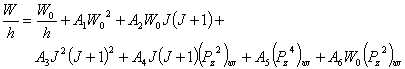
Here, AiˇŻs are constants, W0 is the rigid asymmetric top level energy given above,
![]()
with Izz being the moment of inertia matrix element. In the limit cases of symmetric top or near symmetric top, one has
![]()
Statistic weight
For pure rotational levels gJ = 2J+1. If hyperfine structure splitting exists, each hyperfine structure level has statistic weight of gF = 2F+1.
Selection rules
Selection rules for electric dipole transitions in the absence of external field:
![]()
Electric dipole moment
for transitions
![]()
Here, ¦Ěx is the
permanent dipole moment of the molecule while x is the axis along which the dipole moment is non-zero and so is
responsible for allowed transitions. The calculation of the term ![]() is complex. A table of the values of this term is given for
different x, J, K1, K2,
¦Ę and transition types P, Q, and R in the Appendix V of the book by Townes and
Schawlow (1955). The transition types P, Q and R are corresponding to ¦¤J = -1, 0, and 1, respectively. Here are
several useful relations:
is complex. A table of the values of this term is given for
different x, J, K1, K2,
¦Ę and transition types P, Q, and R in the Appendix V of the book by Townes and
Schawlow (1955). The transition types P, Q and R are corresponding to ¦¤J = -1, 0, and 1, respectively. Here are
several useful relations:

Here x and xˇŻ are the ones with the largest and smallest rotation constant (or momentum of inertia) among the three molecular axes. So the second formula means the change from prolate top base functions to oblate top ones, or vise versa.
For slightly symmetric tops (with parameter ¦Ę close to 1 or -1), the line strength is very close to that of symmetric top by taking Ka as K. One of the good examples of such slightly asymmetric top is SiCC and its isotopologues.
Appendix A
Relative intensity (RI) of
(hyper-)fine structure lines
(from notes by Dr. Dinh-Van-Trung)
The relative intensity of fine or hyperfine structure lines is determined by the quantum mechanic angular momentum coupling theory. First of all, letˇŻs consider a pure rotational transition from any level J to any level JˇŻ, the electric dipole moment of the transition is
![]() .
.
Here, M is the projection quantum number of J along any axis. Therefore, only transition from J to JˇŻ=J-1 has nonzero dipole (allowed). The dipole moment of a transition from one degenerate sublevel of J to all degenerate sublevels of JˇŻ is
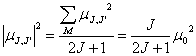 .
.
Hence, line strength for a pure rotation transition is
![]()
Now, letˇŻs consider a HundˇŻs case (b) coupling of rotational angular momentum N, electronic spin S and nuclear spin I. N couples with S to produce angular momentum J, then J couples with I to produce F. This can be expressed using the Dirak bracket as
![]() .
.
Then, the transition dipole moment that is used in the formula of Einstein A coefficient is
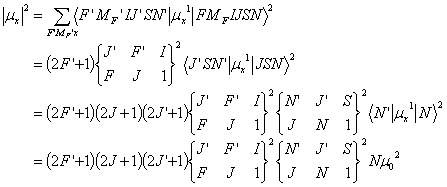
Thus the line strength S can be derived from the relationship:
![]()
Here the symbol {} is the so-called 6-j symbol. The constant ˇ°1ˇ± in the right bottom corner means that we are calculating electric dipole transitions. The formulas to calculate 6-j symbols are
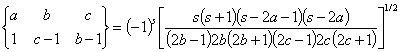
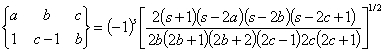
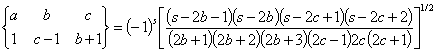
![]()
where s = a+b+c. Following property of the 6-j symbol is important
for its implimentation: exchange of any two columns or exchange of two rows of
any two columns doesnˇŻt change the value. There are more 6-j formulas for other
types of transitions (e.g., quadrupole transtions).
Appendix B
The four HundˇŻs cases for
electronic angular momentum coupling
The angular momenta need to couple with each other because they exert torque on each other, so that some of the momenta have fast precession around other momenta or axes. The fast precession of momenta makes the momenta not conservative any more. But the projection of the fast precession momenta along their precession axes might be still conservative and so can be used to tag the energy levels of the molecule. For a linear molecule with electronic orbital momenta li and spins si, there are four types of most possible coupling cases: the HundˇŻs cases (a,b,c,d). In the four HundˇŻs cases, all individual orbital momenta and spins first couple with each other to form a total orbital momentum L and total spin S. HundˇŻs case (b) is the case found in most astro-molecules. HundˇŻs case (a) is much rarer, while HundˇŻs case (c) and (d) are not found yet.
HundˇŻs case (a) coupling: The coupling between the momenta L, S and the molecular axis is strong. Their projections on the axis are ¦« and ¦˛, respectively. ¦« and ¦˛ add up to form ¦¸. Then ¦¸ couples with couples with rotational angular momentum R to form the resultant angular momentum J. The coupling flow chart:
(1) L+axis=>¦«, S+axis=>¦˛
(2) ¦«+¦˛=>¦¸
(3) ¦¸+R=>J
HundˇŻs case (b) coupling: In this most possible case, the coupling between the momentum L and the molecular axis is still strong, but the coupling of S with the molecular axis is weak. So the projection of L along the axis, ¦«, couples with rotational angular momentum R to form the resultant rotational angular momentum N. N directly couple with S to form the resultant angular momentum J. The coupling flow chart:
(1) L+axis=>¦«
(2) ¦«+R=>N
(3) N+S=>J
HundˇŻs case (c) coupling: In this case, the direct coupling of momenta with molecular axis is weak, L and S first directly couple to resultant electronic angular momentum Jc, then Jc couple with axis with projection ¦¸. ¦¸ further couples with R to form the resultant angular momentum J. The coupling flow chart:
(1) L+S=>Jc
(2) Jc+axis=>¦¸
(3) ¦¸+R=>J
HundˇŻs case (d) coupling: In this case, the coupling of momenta with molecular axis is always very weak, so that L directly couples with R to form resultant angular N, then N directly couples with S to form the resultant angular momentum J. The coupling flow chart:
(1)
L+R=>N
(2)
N+S=>J